Fibrosi cistica
Cause
Quali sono le cause della Fibrosi cistica?
La causa della fibrosi cistica è genetica. La malattia è causata da alterazioni del gene che codifica per la proteina CTFR (regolatore della conduttanza transmembrana della fibrosi cistica) che fa parte di un canale transmembrana per lo ione cloro (Cl-). Un gene difettoso si esprime in una proteina canale CTFR assente o malfunzionante che ostacola il trasporto dello ione cloro attraverso la membrana cellulare.
La proteina CTFR si trova nella membrana apicale delle cellule epiteliali; il polo apicale è la parte funzionale della cellule contrapposto al polo basale, in contiguità con i capillari sanguigni. La proteina CTFR regola gli scambi elettrolitici fra l’interno della cellula e l’ambiente esterno di molte ghiandole dell’organismo.
Il trasporto del cloro ha un ruolo fondamentale nel determinare la consistenza delle secrezioni e lo stato di idratazione degli epiteli. Il gradiente negativo creato dal cloro (Cl-) sulla membrana del polo apicale della cellula epiteliale attrae lo ione sodio (Na+) che, a sua volta, richiama acqua. Il flusso di ione cloro dalla cellula allo spazio extracellulare, anche se non è il diretto responsabile, comporta pertanto il richiamo di acqua nel fluido extracellulare.
In figura è rappresentato il passaggio dello ione cloro (Cl-) (pallini verdi) attraverso la proteina canale CFTR.
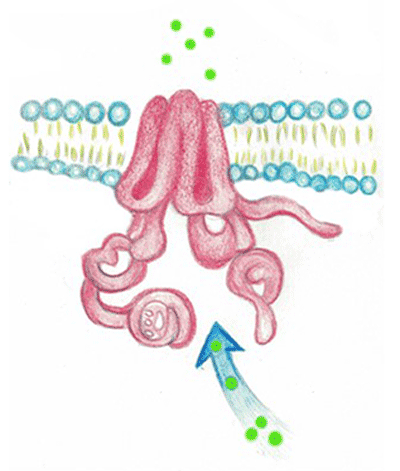
Gli ioni sono strettamente correlati all’acqua, infatti essi sono circondati da una corona di molecole d'acqua di dimensione tanto più grande quanto maggiore è la concentrazione della carica elettrica. Per chiarire questo concetto si possono confrontare due importanti ioni come sodio (Na+) e potassio (K+). Entrambi posseggono la stessa carica elettrica ma, avendo lo ione sodio dimensioni minori, la sua carica elettrica risulta più concentrata, quindi, come si osserva in figura la "nubecola” di molecole di acqua (in azzurro) che lo circonda risulta più grande per il sodio di quella attorno allo ione potassio.

La proteina CTFR è un canale transmembrana ATP-dipendente (Gadsby, Nairm, 1999). Questo vuol dire che spendendo una molecola energetica chiamata ATP (ATP è l’equivalente cellulare del carburante per le automobili) la proteina CFTR è capace di trasportare, contro gradiente di concentrazione (cioè da un ambiente in cui uno ione è meno concentrato a uno in cui è più concentrato), alcuni ioni con carica negativa (anioni), tra cui il cloro.
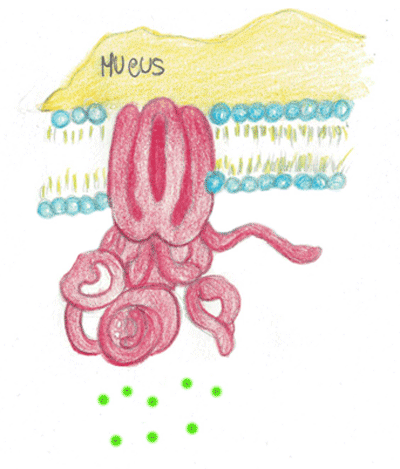
Quando la proteina CTFR è assente o difettosa, il processo che consente di rendere fluido il muco si inceppa e le cellule con funzione ghiandolare esocrina producono secrezioni dense e viscose come ad esempio le ghiandole sudoripare, le ghiandole che producono secrezioni lungo le vie respiratorie, quelle che producono i succhi digestivi e le ghiandole dell'apparato riproduttivo (Ehre et al., 2014).
In figura è rappresentato la proteina CTFR non funzionante e la conseguente formazione di muco denso e viscoso.
La fibrosi cistica si manifesta quando entrambi i geni (uno ricevuto dal padre e uno dalla madre) che codificano per la CTFR sono difettosi (trasmissione autosomica recessiva). Se si eredita una copia del gene funzionante e una difettosa, la fibosi cistica non si manifesta, ma l’individuo sarà portatore sano della malattia e la potrà trasmettare ai figli.
La probabilità di trasmissione alla prole con entrambi i genitori portatori sani del gene CTFR alterato è la seguente:
1 figlio su 4 probabilmente sarà ammalato di fibrosi cistica,
2 figli su 4 probabilmente saranno portatori sani del gene,
1 figlio su 4 probabilmente sarà sano e non porterà la malattia.
Invece la probabilità di trasmissione con un solo genitore portatore sano del gene alterato è:
2 figli su 4 probabilmente saranno portatori sani del gene alterato,
2 figli su 4 probabilmente saranno sani e non potranno trasmettere la malattia.
I difetti genetici noti a carico del gene che codifica per la proteina CTFR sono circa 1900. Questi difetti genetici sono stati suddivisi in 5 gruppi a cui corrispondono manifestazioni della fibrosi cistica più o meno gravi. La mutazione puù frequente (70% dei casi circa) è la mutazione F508del. Nei paesi occidentali circa 1 persona su 25 possiede un gene difettoso per la CTFR. In Italia si stima che i portatori sani, cioè le persone che possiedono solo un gene difettoso, siano circa 2 milioni e mezzo. L’incidenza della malattia nella popolazione caucasica, la più soggetta, è pari a 1 malato ogni 3500 nati (Castellani et al., 2009).
